Molecular Mechanics of Influenza Viral Fusion
Our group is interested in biophysical/-medical problems where mechanical forces play an important role. Among various membrane-related studies, the present project aims at modeling the molecular mechanism of influenza viral fusion.
Influenza
Replication of viruses only occurs after the viral RNA has been delivered to the cell nucleus by merging of the viral and cell membranes. Membrane fusion is thermodynamically favorable, but has a high kinetic barrier. Viral fusion proteins, like the influenza hemagglutinin, deliver the energy required to overcome the barrier and fuse the membranes. Hemagglutinin (HA) is a trimer consisting of 1647 residues that can be divided into two parts: HA1 and HA2. HA1 covers HA2 and is mainly responsible for binding of the virus particle to the cell.
The proposed mechanism of HA-mediated fusion (as illustrated in Figure 1) is as follows: upon acidification HA1 dissociates (Fig. 1A-B) while HA2 changes from its metastable prefusion state into an extended intermediate via a coil-to-helix transition, inserting the amphipathic fusion peptide (red star in the figure) into the target membrane (Fig. 1B-C). A subsequent helix-to-coil transition causes the intermediate to collapse, bringing the two membranes together for fusion (Fig. 1C-E).
This working hypothesis leaves a number of open questions: What are the exact intermediates and what is the kinetics of the transformations between them? We are trying to answer these questions using molecular modeling techniques.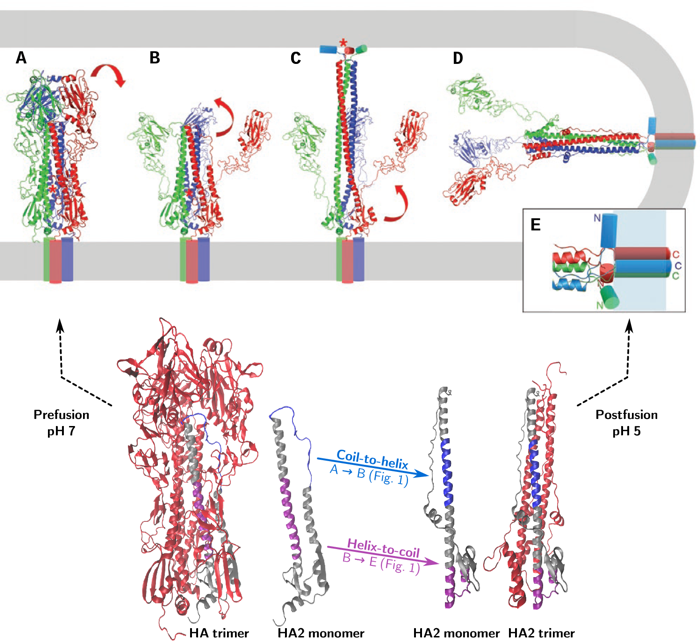
Modeling Hemagglutinin
Direct atomistic simulation of hemagglutinin is computationally intensive because of its size. Therefore, we divide the protein into parts directly related to different steps in the proposed fusion mechanism:
a) Triggering – dissociation of the fusion peptide from its hydrophobic pocket, dissociation of the interface between the HA1 subunits and between HA1 and HA2 (Fig. 1A-B).
b) Formation of the extended intermediate – via a coil-to-helix transition of residues 57 to 75 of HA2 (The ‘B-loop’) (Fig. 1B-C).
c) Collapse of the extended intermediate: helix-to-coil transition of residues 95 to 119 of HA2 (the ‘hinge peptide’) and unfolding of the C-terminal end of HA2 (Fig. 1C-D).
d) Zippering up of the C-terminal end of HA2 along the trimeric coiled coil (Fig. 1C-D).
In a collaborative project with Antoine van Oijen (now at University of Wollongong), we perform all-atom molecular dynamics (MD) simulations to study a number of key events in the formation of the extended conformation and in its subsequence collapse.
Selection of force field and water model
Because we will simulate parts of the protein as separate peptides, special care must be taken in selecting a force field to properly simulate the conformational ensemble of the solvated peptide. The peptides under consideration show helix-to-coil transitions and vice versa, so we need a force field that accurately mimics helical propensities. A recent version of the widely used CHARMM force field, CHARMM36, has been optimized to improve the balance between the sampling of helical and extended conformations. This force field was originally parametrized using a modified version of the TIP3P water model (mTIP3P). Yet, during optimization, the standard TIP3P water model (sTIP3P) has been used. These two water models are already known to yield similar results for globular, stable proteins in their native conformation. However, we have shown that the CHARMM36 force field with mTIP3P water yields less realistic folding than with sTIP3P water [1]. The found combination of force field and water model will be used in our further efforts to elucidate the molecular mechanics of influenza viral fusion.
Key reference:
[1] Boonstra, S., Onck, P.R., van der Giessen, E. (2016).CHARMM TIP3P Water Model Suppresses Peptide Folding by Solvating the Unfolded State. J. Phys. Chem. B, 120 (15), 3692–3698. 10.1021/acs.jpcb.6b01316
| Last modified: | 02 February 2023 12.32 p.m. |