EC. 3.2.1.45
β- Glucosidase = β- Glucocerebrosidase (Gaucher)
De ziekte van Gaucher is een verzamelnaam voor de erfelijke (waarschijnlijk autosomaal recessieve) afwijkingen van het glucosylceramide katabolisme. Bij alle verschijningsvormen van deze ziekte is lysosomaal β- Glucocerebrosidase, ook wel β- Glucosidase genoemd, deficiënt, waardoor een stapeling van glucosylceramide ontstaat. Er worden drie verschillende subtypes van de ziekte van Gaucher onderscheiden die klinisch sterk van elkaar verschillen. Ieder subtype omvat een groep afwijkingen met een gelijksoortig fenotype.
Type I:
Dit type, de chronische niet-neuronopatische variant, komt het vaakst voor, vooral bij midden-Europeaanse Joden. Zowel de leeftijd waarop de ziekte zich openbaart als de ernst van de symptomen kunnen sterk variëren. De eerste verschijnselen bestaan meestal uit splenomegalie en bloedarmoede, later ontstaan hepatomegalie en botafwijkingen, vaak vergezeld van episodes van botpijnen. Gaucher-type I is niet neurologisch, en niet levensbedreigend.
Type II:
Dit type, de acute neuronopatische vorm, heeft meer stereotypische verschijnselen. De ziekte openbaart zich gemiddeld op een leeftijd van drie maanden met hepatosplenomegalie, na zes maanden gevolgd door neurologische afwijkingen. Deze patiënten sterven in het algemeen voor hun tweede verjaardag.
Type III:
Deze variant, de subacute neuronopatische vorm, heeft vergelijkbare klinische verschijnselen als type I en II, waarbij de neurologische afwijkingen zich later openbaren, en minder ernstig zijn.De verschijnselen zijn meer heterogeen, en dienen zich in de jeugd of puberteit aan.
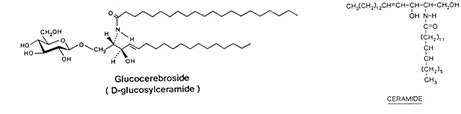
Bij alle drie vormen kan in het weefsel van de patiënten de ‘Gaucher-cel’ gevonden worden. Gaucher-cellen zijn reticulo-endotheliale cellen, waarvan de lysosomen gestapeld zijn met glucosylceramide. Glucosylceramide is samengesteld uit ceramide en glucose (zie figuur). Het ontstaat bij de afbraak van membranen, vooral die van witte bloedcellen.
Normaalgesproken wordt glucosylceramide afgebroken door B -Glucosidase tot ceramide en glucose. Glucosylceramide dat gevonden wordt in milt, lever, nier, plasma en erythrocyten van Gaucher-patiënten, bevat vetzuren met een ketenlengte van ongeveer C22 tot C24.
Glucosylceramide dat gevonden wordt in de hersenen van Gaucher-type II patiënten, bevat voornamelijk C18-vetzuren (stearinezuur).
Principe van de enzymassay:
Het patiëntenmateriaal wordt geincubeerd met het synthetische substraat 4-methylumbelliferyl-β- Glucopyranoside; door de werking van β- Glucosidase wordt 4-methylumbelliferon (4MU) afgesplitst. De hoeveelheid 4MU die zodoende vrijkomt is een maat voor de enzymactiviteit, en fluorometrisch te bepalen.
Benodigd materiaal: leukocyten of fibroblasten.
Referenties:
- Beutler E, Grabowski GA, Gaucher Disease. In:Scriver CR, Beaudet AL, Sly WS, Valle D, eds.The Metabolic and Molecular Bases of Inherited Disease. 2001; 8 edtion: 3635-3669
| Last modified: | 23 February 2015 4.41 p.m. |